int_array = np.array([0,0,0,1,2,3,3,2,1,0,0,1,2,3,2,1,0])
get_peaks(int_array)[(2, 6, 9), (9, 13, 16)]Raw data needs to be preprocessed to simplify it for AlphaPept analysis. Currently, this requires:
Importing raw data frequently results in profile data. When having profile data, Alphapept first needs to perform centroiding to use this data properly. For this, it needs to search local maxima (“peaks”) of the intensity as a function of m/z. For this AlphaPept uses the function get_peaks. A peak is described by three points, the start of the peak, the center, and the end. The function accepts an intensity array and calculates the delta (gradient) between consecutive data points to determine the start, center, and end.
get_peaks (int_array:numpy.ndarray)
Detects peaks in an array.
Args: int_array (np.ndarray): An array with intensity values.
Returns: list: A regular Python list with all peaks. A peak is a triplet of the form (start, center, end)
A quick test to illustrate the function:
int_array = np.array([0,0,0,1,2,3,3,2,1,0,0,1,2,3,2,1,0])
get_peaks(int_array)[(2, 6, 9), (9, 13, 16)]To determine the center of the peak, we distinguish based on the number of raw data points that are contained in the peak:
For the Gaussian estimation, only the three central points are used to fit a Gaussian Peak shape. The Gaussian is then approximated with the logarithm.
The gaussian estimator is defined in gaussian_estimator and is used by the wrapper get_centroid.
gaussian_estimator (peak:tuple, mz_array:numpy.ndarray, int_array:numpy.ndarray)
Three-point gaussian estimator.
Args: peak (tuple): A triplet of the form (start, center, end) mz_array (np.ndarray): An array with mz values. int_array (np.ndarray): An array with intensity values.
Returns: float: The gaussian estimate of the center.
get_centroid (peak:tuple, mz_array:numpy.ndarray, int_array:numpy.ndarray)
Wrapper to estimate centroid center positions.
Args: peak (tuple): A triplet of the form (start, center, end) mz_array (np.ndarray): An array with mz values. int_array (np.ndarray): An array with intensity values.
Returns: tuple: A tuple of the form (center, intensity)
The function performs as expected:
int_array = np.array([0,0,0,1,2,3,3,2,1,0,0,1,2,3,2,1,0])
mz_array = np.array([0,0,1,2,3,4,5,6,7,8,9,10,11,12,13,14,15])
peak = (2, 6, 9)
print(gaussian_estimator(peak, mz_array, int_array))
print(get_centroid(peak, mz_array, int_array))4.499999999999999
(4.499999999999999, 12)The detected centroid can also easily be visualized:
import matplotlib.pyplot as plt
def plot_centroid(peak, mz_array, int_array):
plt.figure(figsize=(3, 3))
start, center, end = peak
centroid = get_centroid((start, center, end), mz_array, int_array)
plt.plot(mz_array[start - 2 : end + 2], int_array[start - 2 : end + 2])
plt.plot(mz_array[start - 2 : end + 2], int_array[start - 2 : end + 2], "b.")
plt.axvline(mz_array[start], color="r")
plt.axvline(mz_array[end], color="r")
plt.axvline(centroid[0], color="g", label='Center')
plt.title("Centroid")
plt.xlabel("m/z")
plt.ylabel("Intensity")
plt.legend()
plt.show()
int_array = np.array([0,0,0,1,2,3,3,2,1,0,0,1,2,3,2,1,0])
mz_array = np.array([0,0,1,2,3,4,5,6,7,8,9,10,11,12,13,14,15])
peak = (2, 6, 9)
plot_centroid(peak, mz_array, int_array)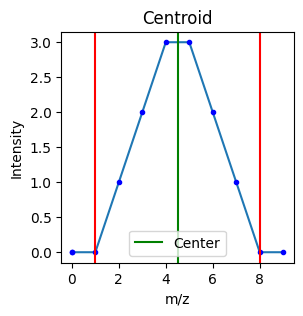
centroid_data (mz_array:numpy.ndarray, int_array:numpy.ndarray)
Estimate centroids and intensities from profile data.
Args: mz_array (np.ndarray): An array with mz values. int_array (np.ndarray): An array with intensity values.
Returns: tuple: A tuple of the form (mz_array_centroided, int_array_centroided)
n most intense peaksget_most_abundant: In order to save spectra in a more memory-efficient form, we only keep the n most abundant peaks. This allows us to save data in a fast, accessible matrix format.
get_local_intensity: This calculates the local intensity to get local maxima.
get_most_abundant (mass:numpy.ndarray, intensity:numpy.ndarray, n_max:int, window:int=10)
Returns the n_max most abundant peaks of a spectrum.
Args: mass (np.ndarray): An array with mz values. intensity (np.ndarray): An array with intensity values. n_max (int): The maximum number of peaks to retain. Setting n_max to -1 returns all peaks. window (int): Use local maximum in a window
Returns: tuple: the filtered mass and intensity arrays.
get_local_intensity (intensity, window=10)
Calculate the local intensity for a spectrum.
Args: intensity (np.ndarray): An array with intensity values. window (int): Window Size Returns: nop.ndarray: local intensity
Input can be read from three different files formats:
.raw files describing Thermo raw data..d folders describing Bruker raw data..mzML files describing generic input..wiff files for SciEx raw data.All reading functions return a query_data dictionary similar to the following structure:
query_data = {
"scan_list_ms1": np.array(...)
"rt_list_ms1": np.array(...)
"mass_list_ms1": np.array(...)
"int_list_ms1": np.array(...)
"ms_list_ms1": np.array(...)
"scan_list_ms2": np.array(...)
"rt_list_ms2": np.array(...)
"mass_list_ms2": mass_list_ms2
"int_list_ms2": int_list_ms2
"ms_list_ms2": np.array(...)
"prec_mass_list2": np.array(...)
"mono_mzs2": np.array(...)
"charge2": np.array(...)
}To read Thermo files, AlphaPept uses the pyrawfilereader package, a Python implementation of the commonly used rawfilereader tool. By using the custom python version, Thermo files can be read without having to install MSFileReader.
The user can pass an additional flag use_profile_ms1. This will then use the profile data which is not centroided already an peform centroiding. Note that this will lead to slightly different intensities, as the centroided data uses the apex and the centroid algorithm the summed intensity.
The import of files is done now via alpharaw.
load_thermo_raw (raw_file_name:str, n_most_abundant:int, use_profile_ms1:bool=False, callback:<built- infunctioncallable>=None)
Load raw thermo data as a dictionary.
Args: raw_file_name (str): The name of a Thermo .raw file. n_most_abundant (int): The maximum number of peaks to retain per MS2 spectrum. use_profile_ms1 (bool): Use profile data or centroid it beforehand. Defaults to False. callback (callable): A function that accepts a float between 0 and 1 as progress. Defaults to None.
Returns: tuple: A dictionary with all the raw data and a string with the acquisition_date_time
To access Bruker files, AlphaPept relies on the external timsdata library from Bruker (available in the alphatims\ext folder, licenses are applicable). Unfortunately, these libraries are only available on Windows and Linux. As a result, the reading of raw data is not available on macOS. However, once raw data is converted to .ms_data.hdf output, other workflow steps (besides feature feating) are possible without problems on macOS.
load_bruker_raw (raw_file, most_abundant, callback=None, **kwargs)
Load bruker raw file and extract spectra
For ccs (i.e., ion mobility) values, we need additional functions from the Bruker library. As the live feature-finder might not be able to determine some charge values, it is intended to perform this calculation at a later stage once we have charge values from the post-processing feature finder.
one_over_k0_to_CCS (one_over_k0s:numpy.ndarray, charges:numpy.ndarray, mzs:numpy.ndarray)
Retrieve collisional cross section (CCS) values from (mobility, charge, mz) arrays.
Args: one_over_k0s (np.ndarray): The ion mobilities (1D-np.float). charges (np.ndarray): The charges (1D-np.int). mzs (np.ndarray): The mz values (1D-np.float).
Returns: np.ndarray: The CCS values.
Due to availbility of Bruker libraries, this can only be tested on Windows and Linux.
.wiff filesSimilar to Thermo, we now use alpharaw to read .wiff-files.
load_sciex_raw (raw_file_name:str, n_most_abundant:int, callback:<built- infunctioncallable>=None)
Load Sciex data as a dictionary.
Args: raw_file_name (str): The name of a Sciex .wiff file. n_most_abundant (int): The maximum number of peaks to retain per MS2 spectrum. use_profile_ms1 (bool): Use profile data or centroid it beforehand. Defaults to False. callback (callable): A function that accepts a float between 0 and 1 as progress. Defaults to None.
Returns: tuple: A dictionary with all the raw data and a string with the acquisition_date_time
Note: SciEx file have one main file but multiple additional files which need to be present in the same location.
import_sciex_as_alphapept (sciex_wiff)
.mzML filesTo access .mzML files, we rely on the pyteomics package. For using an mzml format for performing a search, Peak Picking (data centroiding) should be applied to all MS levels of the data.
load_mzml_data (filename:str, n_most_abundant:int, callback:<built- infunctioncallable>=None, **kwargs)
Load data from an mzml file as a dictionary.
Args: filename (str): The name of a .mzml file. n_most_abundant (int): The maximum number of peaks to retain per MS2 spectrum. callback (callable): A function that accepts a float between 0 and 1 as progress. Defaults to None.
Returns: tuple: A dictionary with all the raw data, a string with the acquisition_date_time and a string with the vendor.
extract_mzml_info (input_dict:dict)
Extract basic MS coordinate arrays from a dictionary.
Args: input_dict (dict): A dictionary obtained by iterating over a Pyteomics mzml.read function.
Returns: tuple: The rt, masses, intensities, ms_order, prec_mass, mono_mz, charge arrays retrieved from the input_dict. If the ms level in the input dict does not equal 2, the charge, mono_mz and prec_mass will be equal to 0.
check_sanity (mass_list:numpy.ndarray)
Sanity check for mass list to make sure the masses are sorted.
Args: mass_list (np.ndarray): The mz values (1D-np.float).
Raises: ValueError: When the mz values are not sorted.
Benchmarking proteomics software against each other is not straightforward as various naming conventions exist and different algorithms are implemented. In this section, we define some helper functions that allow us to read results from other tools and facilitate the comparison of different tools against AlphaPept.
One of the most commonly used tools to analyze MS data is MaxQuant. AlphaPept reads MaxQuant .xml files as follows:
extract_mq_settings (path:str)
Function to return MaxQuant values as a dictionary for a given xml file.
Args: path (str): File name of an xml file.
Returns: dict: A dictionary with MaxQuant info.
Raises: ValueError: When path is not a valid xml file.
mq_dict = extract_mq_settings('../testfiles/test_mqpar.xml')
mq_dict['fastaFiles']{'FastaFileInfo': {'fastaFilePath': 'testfile.fasta',
'identifierParseRule': '>([^\\s]*)',
'descriptionParseRule': '>(.*)',
'taxonomyParseRule': None,
'variationParseRule': None,
'modificationParseRule': None,
'taxonomyId': None}}AlphaPept incorporates PTMs directly in amino acid sequences with lower case identifiers. Parsing MaxQuant sequences with PTMs is done with:
parse_mq_seq (peptide:str)
Replaces maxquant convention to alphapept convention.
ToDo: include more sequences
Args: peptide (str): A peptide sequence from MaxQuant.
Returns: str: A parsed peptide sequence compatible with AlphaPept.
A quick test shows the results are correct:
parse_mq_seq('_AFQPFFVELTM(Oxidation (M))PYSVIR_')'AFQPFFVELToxMPYSVIR'For saving, we are currently relying on the hdf-container (see below).
While we could, in principle, store the mz and int arrays as a list of variable length, this will come at a performance decrease. We, therefore, create an array of the dimensions of the n most abundant peaks and the number of spectra with the function list_to_numpy_f32 and fill the unoccupied cells with -1. This allows an increase in accessing times at the cost of additional disk space.
list_to_numpy_f32 (long_list:list)
Function to convert a list to np.float32 array.
Args: long_list (list): A regular Python list with values that can be converted to floats.
Returns: np.ndarray: A np.float32 array.
As MS hardware has continued to improve over the years, MS data has become more complex. To deal with this complexity, the MS community has already used many different data formats to store and access data. HDF containers are one option, but they have not yet gained widespread support.
In general, an HDF container can be viewed as a compressed folder with metadata (i.e., attributes) associated to each single subfolder or file (i.e., data arrays of various types and sizes) within this container. A container might, for instance have contents that look like, e.g.:
HDF_Container
{
meta_data_1: "Some string",
meta_data_2: 1234567890,
...
}
array_1
{
meta_data_of_array1_1: "Some other string",
...
},
100x2 int8
array_2
1000x2 float64
subfolder_1
{
meta_data_of_subfolder_1_1: "Really any string of any length",
...
}
array_1_of_subfolder_1
subfolder_1_1
...
subfolder_n
...A few of the advantages of HDF are, e.g.:
For these reasons, HDF containers have gained popularity in several scientific fields, including (astro)physics and geology. It is, therefore, no surprise that Python has excellent support for HDF containers. The two most used packages are h5py and tables, where the former has a generic API and the second is frequently used with pandas dataframes. An excellent viewer for HDF files is HDF Compass.
AlphaPept uses the python h5py package to store MS data in HDF containers, inspired by the ion_networks repository.
creation time, original_file_name and version.file_name of an HDF container, an is_read_only flag, an is_overwritable flag and is_new_file flag.last_updated and a check function to warn users about potential compatibility issues.HDF_File (file_name:str, is_read_only:bool=True, is_new_file:bool=False, is_overwritable:bool=False)
A generic class to store and retrieve on-disk data with an HDF container.
Contents of HDF containers come in three variants:
Groups: foldersDatasets: arraysAttributes: metadata associated with individual datasets or groups (with the root folder also considered as a normal group)These contents can be accessed with read and write functions.
HDF_File.write (value, group_name:str=None, dataset_name:str=None, attr_name:str=None, overwrite:bool=None, dataset_compression:str=None, swmr:bool=False)
Write a value to an HDF_File.
Args: value (type): The name of the data to write. If the value is pd.DataFrame, a dataset_name must be provided. group_name (str): The group where to write data. If no group_name is provided, write directly to the root group. Defaults to None. dataset_name (str): If no dataset_name is provided, create a new group with value as name. The dataset where to write data. Defaults to None. attr_name (str): The attr where to write data. Defaults to None. overwrite (bool): Overwrite pre-existing data and truncate existing groups. If the False, ignore the is_overwritable flag of this HDF_File. Defaults to None. dataset_compression (str): The compression type to use for datasets. Defaults to None. swmr (bool): Open files in swmr mode. Defaults to False.
Raises: IOError: When the object is read-only. KeyError: When the group_name or attr_name does not exist. ValueError: When trying to overwrite something while overwiting is disabled.
HDF_File.read (group_name:str=None, dataset_name:str=None, attr_name:str=None, return_dataset_shape:bool=False, return_dataset_dtype:bool=False, return_dataset_slice:slice=slice(None, None, None), swmr:bool=False)
Read contents of an HDF_File.
Args: group_name (str): The group_name from where to read data. If no group_name has been provided, read directly from the root group. Defaults to None. dataset_name (str): The dataset to read. If no dataset_name has been provided, read directly from the group. If the dataset_name refers to a group, it is assumed to be pd.DataFrame and returned as such. Defaults to None. attr_name (str): The attribute to read. If attr_name is not None, read the attribute value instead of the contents of a group or dataset. If attr_name == ““, read all attributes as a dict. Defaults to None. return_dataset_shape (bool): Do not read complete dataset to minimize RAM and IO usage. Defaults to False. return_dataset_dtype (bool): Do not read complete dataset to minimize RAM and IO usage. Defaults to False. return_dataset_slice (slice): Do not read complete dataset to minimize RAM and IO usage. Defaults to slice(None). swmr (bool): Use swmr mode to read data. Defaults to False.
Returns: type: Depending on what is requested, a dict, value, np.ndarray or pd.dataframe is returned.
Raises: KeyError: When the group_name does not exist. KeyError: When the attr_name does not exist in the group or dataset. KeyError: When the dataset_name does not exist in the group. ValueError: When the requested dataset is not a np.ndarray or pd.dataframe.
Unit tests for this generic HDF class include:
.ms_data.hdf fileBased on the generic HDF_File, a subclass that acts as an MS data container can be implemented. This class should contain all (centroided) fragment ions, all their coordinates, and all the metadata.
MS_Data_File (file_name:str, is_read_only:bool=True, is_new_file:bool=False, is_overwritable:bool=False)
A class to store and retrieve on-disk MS data with an HDF container.
A single generic function should allow to read raw data and store spectra. Different arguments allow different vendor formats.
index_ragged_list (ragged_list:list)
Create lookup indices for a list of arrays for concatenation.
Args: value (list): Input list of arrays.
Returns: indices: A numpy array with indices.
MS_Data_File.import_raw_DDA_data (file_name:str, n_most_abundant:int=-1, callback:<built- infunctioncallable>=None, query_data:dict=None, vendor:str=None)
Load centroided data and save it to this object.
Args: file_name (str): The file name with raw data (Thermo, Bruker or mzml). n_most_abundant (int): The maximum number of peaks to retain per MS2 spectrum. Defaults to -1. callback (callable): A function that accepts a float between 0 and 1 as progress. Defaults to None. query_data (dict): A dictionary with raw data. If this is not None, data will only be saved and not imported. Defaults to None. vendor (str): The vendor name, must be Thermo or Bruker if provided. Defaults to None.
Testing of the MS_Data_File container includes reading and writing from different file formats.
While that HDF data structure could be used directly, it is often easier to read it and return a query_data dictionary similar to those that are returned by the readers of Thermo, Bruker, mzML and mzXML raw data.
MS_Data_File.read_DDA_query_data (calibrated_fragments:bool=False, force_recalibrate:bool=False, swmr:bool=False, **kwargs)
Read query data from this ms_data object and return it as a query_dict.
Args: calibrated_fragments (bool): If True, calibrated fragments are retrieved. Calibration offsets can already be present in the ms_data or recalculated. Defaults to False. force_recalibrate (bool): If calibrated fragments is True, recalibrate mzs values even if a recalibration is already provided. Defaults to False. swmr (bool): Open the file in swmr mode. Defaults to False. **kwargs (type): Can contain a database file name that was used for recalibration.
Returns: dict: A query_dict with data for MS1 and MS2 scans.
To use all the above functionality from a workflow with several parameters, the following functions are defined. These functions also allow parallel processing.
raw_conversion (to_process:dict, callback:<built- infunctioncallable>=None, parallel:bool=False)
Wrapper function to convert raw to ms_data_file hdf.
Args: to_process (dict): A dictionary with settings indicating which files are to be processed and how. callback (callable): A function that accepts a float between 0 and 1 as progress. Defaults to None. parallel (bool): If True, process multiple files in parallel. This is not implemented yet! Defaults to False.
Returns: bool: True if and only if the conversion was succesful.
The following commands are bookkeeping to make sure this and other notebooks are properly parsed to python modules.