Analysis of PELSA peptidomics data (Li et al, 2025)#
Study background#
PELSA [1] is a novel proteomic method to investigate protein-ligand interactions through limited proteolysis. In a nutshell, proteins in solution (typically a cellular lysate) are briefly (typically between 1 and 4 minutes, depending on the specific protocol) treated with the protease trypsin at very high enzyme : substrate ratio (typically 1:2). Trypsin cuts the protein at K and R residues, but digesting very briefly means that only surface exposed K/R sites are actually cut. The critical assumption is that, if a ligand (such as a small molecule) is bound to the protein surface, it stablizes the surrounding protein region and digestion is momentarily slowed. It is worth noting that this is not strictly confined to actual steric blockage, but stablizing effects that extend beyond the ligand’s footprint. The key to PELSA is that, when a ligand is present, K/R sites around the binding site are cut at slower rate than if the ligand is not present. If digestion is allowed to proceed for too long, this dynamic effect vanishes as the entire protein gets digested. However, the short digestion time means that peptides which are dynamically stabilized (i.e. ligand-adjacent) will appear as less abundant than their unbound control counterparts. We replicate the original publication’s analysis of samples treated with Staurosporine, a pan-kinase binder, and its comparison against DMSO treated samples and visualize the regulation of kinase targets.
Key proteomics methods and considerations#
Challenge |
Why It Matters |
Our Approach |
|---|---|---|
Missing values |
Proteomics data is ~20-50% missing (MNAR) |
Evaluate whether imputation strategies improve results |
Dynamic range |
Precursors span 6+ orders of magnitude |
Log-transform intensities |
Data layers |
Measurement is in precursors, but readout of regulated proteins is preferred |
Evaluate published aggregation on UniProt ID and evaluate aggregation directly on gene name |
References#
[1]: Li, Kejia, et al. “A peptide-centric local stability assay enables proteome-scale identification of the protein targets and binding regions of diverse ligands.” Nature Methods 22.2 (2025): 278-282.
[2]: Ammar, Constantin, et al. “Tree-based quantification infers proteoform regulation in bottom-up proteomics data.” bioRxiv (2025): 2025-03.
[3]: SmythLab/limpa https://github.com/SmythLab/limpa
[4]: Li, Kejia, et al. “High-throughput peptide-centric local stability assay extends protein-ligand identification to membrane proteins, tissues, and bacteria.” bioRxiv (2025): 2025-04.
Analysis#
%load_ext autoreload
%autoreload 2
import tempfile
import pandas as pd
import numpy as np
import logging
import alphapepttools as apt
from matplotlib.patches import Rectangle
logger = logging.getLogger(__name__)
1. Data loading#
1.1. Preparing the dataset using alphapepttools loaders and AnnData factory.#
For this study, the relevant files were obtained from study’s PRIDE-repository (https://www.ebi.ac.uk/pride/archive/projects/PXD034606) and processed with DIANN 2.1.0. and a human spectral library. The resulting long-format precursor table is available through alphapepttools’ data module. In addition, corresponding sample metadata has been added to differentiate Staurosporine-treated from control samples. Below is the list of .raw files which were processed for this notebook:
LKJ_20211007_480_Hela_stau_0uM_1.raw
LKJ_20211007_480_Hela_stau_0uM_2.raw
LKJ_20211007_480_Hela_stau_0uM_3.raw
LKJ_20211007_480_Hela_stau_0uM_4.raw
LKJ_20211007_480_Hela_stau_20uM_1.raw
LKJ_20211007_480_Hela_stau_20uM_2.raw
LKJ_20211007_480_Hela_stau_20uM_3.raw
LKJ_20211007_480_Hela_stau_20uM_4.raw
We have to extract protein-level intensities and precursor-level intensities from this dataset using the alphapepttools.io.AnnDataFactory class. The resulting AnnData object contains protein-group or precursor quantities and any number of feature-metadata columns (for example, protein groups may have genes as secondary annotation, precursors may have protein groups and genes as secondary annotation). Note that the names we use below to refere to columns are not actual DIANN fields. read_psm_table runs alphabase’s loaders under the hood, which standardize column names and sequence annotation syntax across all supported search engines.
# Download the data using the alphapepttools data module
report_path = apt.data.get_data("pelsa_report_diann", output_dir=tempfile.mkdtemp())
sample_metadata_path = apt.data.get_data("pelsa_metadata", output_dir=tempfile.mkdtemp())
# Get the full report
full_report = pd.read_parquet(report_path)
# AnnDataFactory instance containing precursor level data
# Note that Protein.Ids is not a standardized alphabase column; since we need it for this experiment, we can specify it as a var-column to be retained in the resulting AnnDataFactory object
adata_precursor = apt.io.read_psm_table(
file_paths=report_path,
search_engine="diann",
level="precursors",
var_columns=["proteins", "genes", "sequence"],
)
# AnnDataFactory instance containing protein level data
adata_protein = apt.io.read_psm_table(
file_paths=report_path,
search_engine="diann",
level="proteins",
var_columns=["genes"],
)
# AnnDataFactory instance containing gene level data
adata_gene = apt.io.read_psm_table(
file_paths=report_path,
search_engine="diann",
level="genes",
var_columns=["genes"],
)
/var/folders/2l/hhd_z4hx3070zw8rlj4c3l940000gn/T/tmpban0s88j/report.parquet does not yet exist
/var/folders/2l/hhd_z4hx3070zw8rlj4c3l940000gn/T/tmpban0s88j/report.parquet successfully downloaded (70.8467435836792 MB)
/var/folders/2l/hhd_z4hx3070zw8rlj4c3l940000gn/T/tmp46rc07xv/iteration_L_literature_reprocessing_PELSA_samplemap.csv does not yet exist
/var/folders/2l/hhd_z4hx3070zw8rlj4c3l940000gn/T/tmp46rc07xv/iteration_L_literature_reprocessing_PELSA_samplemap.csv successfully downloaded (0.000179290771484375 MB)
1.2. Adding sample metadata#
We focus on Protein Groups and Precursors, and use pp.add_metadata() to merge the metadata into our AnnData instances. Note that add_metadata does not just concatenate shape-matched tables to data, but checks for matching indices. The advantage of this is that the same AnnData instance can be reannotated with multiple metadata tables which don’t have to match the data in shape or order.
Tip: Since the matching index is a prerequisite, it is sometimes helpful to map actual file names to metadata by a mapping dictionary as shown below. Generally, it is always preferred to set up metadata such that individual samples are already identified by their rawfile names, but in practice this is often not the case, necessitating a mapping step on some unique sample ID, derived from the rawfile name on the data side, and from the metadata columns on the metadata side.
adata_precursor.to_df()
| precursor_id | (UniMod:1)AAAAAAAGDSDSWDADAFSVEDPVR3 | (UniMod:1)AAAAAAAGDSDSWDADAFSVEDPVRK3 | (UniMod:1)AAAAAAAPSGGGGGGEEERLEEK3 | (UniMod:1)AAAAAAGAASGLPGPVAQGLK3 | (UniMod:1)AAAAAAGEAR2 | (UniMod:1)AAAAAAGPSPGSGPGDSPEGPEGEAPER3 | (UniMod:1)AAAAAAGPSPGSGPGDSPEGPEGEAPERR3 | (UniMod:1)AAAAAAGSGTPREEEGPAGEAAASQPQAPTSVPGAR4 | (UniMod:1)AAAAAETPEVLR2 | (UniMod:1)AAAAAGLGGGGAGPGPEAGDFLAR3 | ... | YYVSPSDLLDDKTASPIILHENNVSR4 | YYVTIIDAPGHR2 | YYVTIIDAPGHR3 | YYVTIIDAPGHRDFIK3 | YYVTIIDAPGHRDFIK4 | YYWDEYK2 | YYYIPQYK2 | YYYQLNSK2 | YYYSDNFFDGQR2 | YYYVQNVYTPVDEHVYPDHR3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| raw_name | |||||||||||||||||||||
| LKJ_20211007_480_Hela_stau_0uM_1 | 21011194.0 | 9.989504e+05 | 1204468.875 | 8206635.5 | 4197046.00 | 2899150.00 | 5279800.5 | 530959.3750 | 3692616.750 | 18911396.0 | ... | 4178915.0 | 381038432.0 | 2.454794e+09 | 2738576.75 | 26054414.0 | 369964.3125 | 17009144.0 | 10044290.0 | 4289190.00 | 12370400.0 |
| LKJ_20211007_480_Hela_stau_0uM_2 | 39567784.0 | 1.193255e+06 | 1113355.375 | 7573107.5 | 2290991.25 | 2400930.50 | 5289624.5 | 525287.0000 | 2618211.500 | NaN | ... | 4506887.0 | 289599392.0 | 2.901842e+09 | 3140537.75 | 36270848.0 | 262792.8750 | 14329460.0 | 13048745.0 | 5184251.00 | 11958440.0 |
| LKJ_20211007_480_Hela_stau_0uM_3 | 19115586.0 | 1.804508e+06 | 1456042.625 | 8628502.0 | 4230691.00 | 2561574.25 | 4938099.0 | 533274.6250 | 3216539.750 | 21216458.0 | ... | 5838578.0 | 296343072.0 | 2.462786e+09 | 3446117.50 | 32477234.0 | NaN | 9924278.0 | 11247496.0 | 3635518.25 | 11344828.0 |
| LKJ_20211007_480_Hela_stau_0uM_4 | 26157658.0 | 1.432018e+06 | 1232615.625 | 8755009.0 | 2545534.50 | 2966516.25 | 6838089.0 | 871199.2500 | 3554148.750 | 22884012.0 | ... | 5823588.0 | 316219744.0 | 2.778846e+09 | 3197354.00 | 34685588.0 | 519398.0625 | 6488640.5 | 12535207.0 | 4060651.00 | 11446639.0 |
| LKJ_20211007_480_Hela_stau_20uM_1 | 16399891.0 | 1.071585e+06 | 1422827.250 | 8666873.0 | 3098686.00 | 3243728.25 | 7012452.5 | 827002.1250 | 2701790.250 | 22141386.0 | ... | 4799338.0 | 307733792.0 | 2.803580e+09 | 4438438.00 | 38047740.0 | NaN | 13708995.0 | 10929723.0 | 3247188.75 | 11592111.0 |
| LKJ_20211007_480_Hela_stau_20uM_2 | 15837731.0 | 1.638444e+06 | 1449917.250 | 7008343.0 | 3313300.50 | 3245684.25 | 7857120.0 | 847267.5000 | 3120052.750 | 22378316.0 | ... | 4583105.0 | 292125696.0 | 2.398915e+09 | 4048503.75 | 34615580.0 | NaN | 11574317.0 | 13034708.0 | 5064767.00 | 11844795.0 |
| LKJ_20211007_480_Hela_stau_20uM_3 | 11208110.0 | 9.845828e+05 | 1306324.625 | 9122322.0 | 3581558.75 | 3032285.25 | 7944031.0 | 907395.9375 | 1991426.125 | 20551044.0 | ... | 5581498.0 | 296175680.0 | 2.226232e+09 | 3393078.00 | 32654838.0 | NaN | NaN | 11486361.0 | 4304476.50 | 10728446.0 |
| LKJ_20211007_480_Hela_stau_20uM_4 | 16460328.0 | 1.132030e+06 | 1284206.500 | 8428735.0 | 3931314.75 | 3631191.50 | 8004403.0 | 898860.4375 | 3040418.250 | 20938560.0 | ... | 5675199.5 | 213132960.0 | 2.313531e+09 | 3534630.25 | 28262632.0 | NaN | 11812809.0 | 12613163.0 | 4454296.00 | 13792679.0 |
8 rows × 91803 columns
sample_metadata = pd.read_csv(sample_metadata_path)
display(sample_metadata)
# Create a unique identifier for each row in the sample metadata by combining the plate and running_count columns
sample_metadata["sample"] = sample_metadata["plate"] + "_" + sample_metadata["running_count"].astype(str)
# Create a mapping from the unique identifier to the original index in the adata_protein.obs_names
run_name_map = {"_".join(k.split("_")[-2:]): k for k in adata_protein.obs_names}
print(run_name_map)
# Replace the index of the sample metadata with the unique identifier and map it to the original index in adata_protein.obs_names
sample_metadata.index = sample_metadata["sample"].map(run_name_map)
| running_count | plate | treatment | instrument | |
|---|---|---|---|---|
| 0 | 1 | 0uM | CTRL | E480 |
| 1 | 2 | 0uM | CTRL | E480 |
| 2 | 3 | 0uM | CTRL | E480 |
| 3 | 4 | 0uM | CTRL | E480 |
| 4 | 1 | 20uM | Stauro | E480 |
| 5 | 2 | 20uM | Stauro | E480 |
| 6 | 3 | 20uM | Stauro | E480 |
| 7 | 4 | 20uM | Stauro | E480 |
{'0uM_1': 'LKJ_20211007_480_Hela_stau_0uM_1', '0uM_2': 'LKJ_20211007_480_Hela_stau_0uM_2', '0uM_3': 'LKJ_20211007_480_Hela_stau_0uM_3', '0uM_4': 'LKJ_20211007_480_Hela_stau_0uM_4', '20uM_1': 'LKJ_20211007_480_Hela_stau_20uM_1', '20uM_2': 'LKJ_20211007_480_Hela_stau_20uM_2', '20uM_3': 'LKJ_20211007_480_Hela_stau_20uM_3', '20uM_4': 'LKJ_20211007_480_Hela_stau_20uM_4'}
# Add the metadata to all AnnData instances
adata_precursor = apt.pp.add_metadata(adata_precursor, sample_metadata, axis=0)
adata_protein = apt.pp.add_metadata(adata_protein, sample_metadata, axis=0)
adata_gene = apt.pp.add_metadata(adata_gene, sample_metadata, axis=0)
1.3. Log2-transformation of proteomics data#
We log2-transform our data, but keep the untransformed values in a separate layer called ‘raw’. Due to underlying averaging operations, calculations like Coefficient of Variation (CV) should always be done on the non-log-transformed values (computing the mean of logged values carries different statistical implications than taking the mean of untransformed values).
# First, move the original data to a new layer called "raw" to preserve it before log transformation
adata_precursor.layers["raw"] = adata_precursor.X.copy()
adata_protein.layers["raw"] = adata_protein.X.copy()
adata_gene.layers["raw"] = adata_gene.X.copy()
# Apply log-transformation. Note that the nanlog function automatically handles zeros and other invalid values, and prints a small summary to inform about special values encountered during the transformation.
apt.pp.nanlog(adata_precursor)
apt.pp.nanlog(adata_protein)
apt.pp.nanlog(adata_gene)
# Inspect the log2-transformed values:
display(adata_protein.to_df().iloc[:5, :5])
# Raw values:
display(adata_protein.to_df(layer="raw").iloc[:5, :5])
| proteins | A0A096LP55;P07919 | A0A0B4J2F0 | A0A1B0GUY1 | A0A3B3IS91 | A0A3B3IU46;Q9BTL3 |
|---|---|---|---|---|---|
| raw_name | |||||
| LKJ_20211007_480_Hela_stau_0uM_1 | 21.790016 | 23.018673 | 24.692411 | 17.029541 | 23.745449 |
| LKJ_20211007_480_Hela_stau_0uM_2 | 21.902254 | 23.051476 | 24.746101 | 17.110256 | 23.861660 |
| LKJ_20211007_480_Hela_stau_0uM_3 | 21.277990 | 23.003662 | 24.555399 | 17.807335 | 23.645226 |
| LKJ_20211007_480_Hela_stau_0uM_4 | 21.540596 | 23.048594 | 24.787121 | 16.149979 | 23.457727 |
| LKJ_20211007_480_Hela_stau_20uM_1 | 21.918283 | 23.059902 | 24.633326 | NaN | 23.821888 |
| proteins | A0A096LP55;P07919 | A0A0B4J2F0 | A0A1B0GUY1 | A0A3B3IS91 | A0A3B3IU46;Q9BTL3 |
|---|---|---|---|---|---|
| raw_name | |||||
| LKJ_20211007_480_Hela_stau_0uM_1 | 3626171.50 | 8497893.0 | 27111686.0 | 133783.515625 | 14063468.0 |
| LKJ_20211007_480_Hela_stau_0uM_2 | 3919543.50 | 8693320.0 | 28139656.0 | 141481.781250 | 15243168.0 |
| LKJ_20211007_480_Hela_stau_0uM_3 | 2542808.25 | 8409926.0 | 24655366.0 | 229372.937500 | 13119653.0 |
| LKJ_20211007_480_Hela_stau_0uM_4 | 3050461.00 | 8675965.0 | 28951204.0 | 72715.656250 | 11520718.0 |
| LKJ_20211007_480_Hela_stau_20uM_1 | 3963336.50 | 8744247.0 | 26023742.0 | NaN | 14828694.0 |
2. Perform basic EDA on protein and precursor data#
2.1. Coefficient of Variation analysis#
In proteomics, we commonly compare different sample groups using significance tests such as (but not limited to) the two-sided, two-sample independent Student’s t-test. It gives us a t-value, which can be interpreted as a standardized fold change between sample groups - standardized, because a normalization for sample-size and variance is baked into the formula.
with the pooled standard deviation \(s_p\) given as
The resulting t-value, plus a known t-distribution are used to compute the p-value, which is commonly interpreted as “the probability of seeing a difference as extreme or more extreme than the one we see, if we assume that both sample groups come from the same underlying population”. A common threshold for significance is set at 0.05, though applications of thresholds vary based on the actual experiment and whether it is more costly to have false-negatives or false-positives.
This means that having high intra-group variance, i.e. high variation within the group directly reduces the power of our t-test.
Therefore, we must check whether intra-group variance levels are acceptable, commonly done by assessing the Coefficient of Variation or CV:
where \(\sigma\) or \(s\) denote the population or sample (in practice mostly sample) standard deviation and \(\mu\) or \(\bar{x}\) denote the population or sample (in practice mostly sample) mean.
Note that it is critical to compute CVs on actual intensities, not log-transformed values. Log-transformation breaks the proportional relationship between mean and standard deviation by effectively stabilizing the standard deviation, which causes the SD/mean ratio to shrink as the mean grows, resulting in misleading CVs*
* For example, the two groups [80, 100, 120] and [8000, 10000, 12000] have means of 100 and 10000, standard deviations of 20 and 2000 and therefore CVs of 20 % and 20 %, respectively. If we log2-transform the values to [6.322, 6.644, 6.907] and [12.966, 13.288, 13.551], the respective means become 6.624 and 13.268, the sample standard deviations become 0.293 and 0.293, and therefore the respective CVs are 0.044 and 0.022 –> the second group appears as more “precise” just from log-transforming.
Since PELSA-assay precursor fold changes are generally smaller than e.g. pulldowns, it is especially important to control the spread of replicates. In other words, too high CVs will drown out the signal and cause high numbers of non-significant precursor fold-changes.
We run AlphaPeptTools’ coefficient_of_variation function with specified sample groups to get the CVs of both treatments**
adata_protein = apt.metrics.coefficient_of_variation(
adata=adata_protein,
group_column="treatment",
key_added="cv_treatment",
layer="raw",
copy=True,
min_valid=2,
)
adata_precursor = apt.metrics.coefficient_of_variation(
adata=adata_precursor,
group_column="treatment",
key_added="cv_treatment",
layer="raw",
copy=True,
min_valid=2,
)
Note: When there are multiple sample groups to compute CVs on, the coefficient_of_variation function adds the results to AnnData’s .varm dictionary. The advantage of this approach is that .var does not get polluted with many different, sample-group dependent column names. Instead, the columns of the dataframe stored in .varm correspond to the actual sample groups, and we can get the whole dataframe containing the CVs back by simply calling adata.varm[<key>], where <key>means the key_added argument of coefficient_of_variation, which defaults to cv.
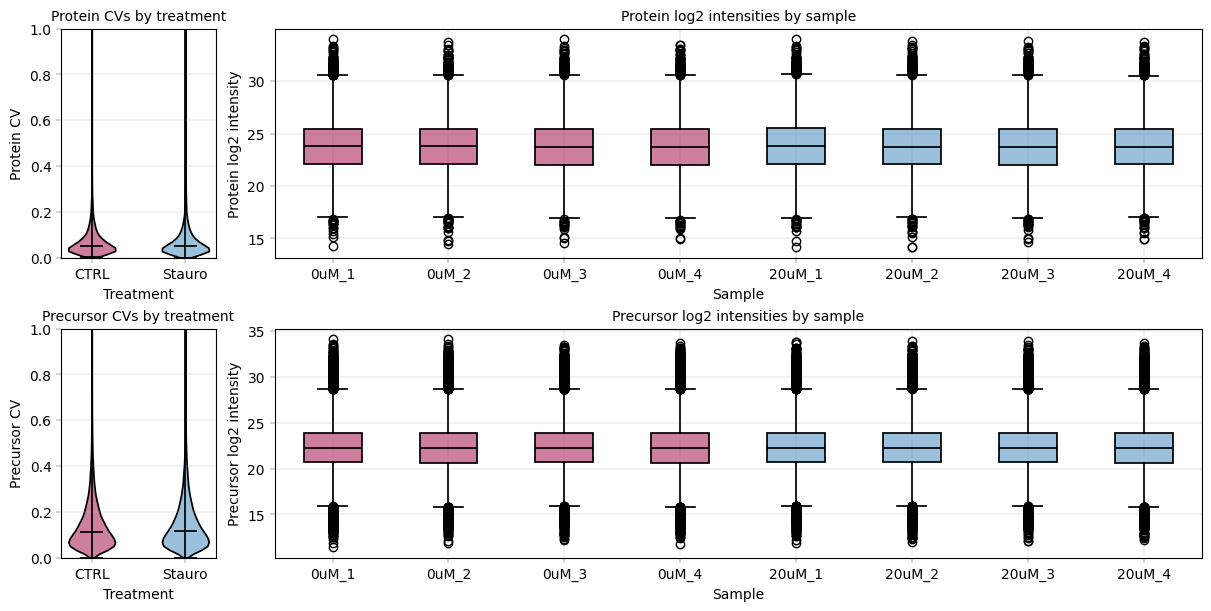
We use alphapepttools’ plotting syntax to generate a stylized panel with violin plots for sample group feature CVs, and to assess the median intensity of all features across samples.
# Generate an AxisManager object ('axm') which is essentially matplotlib subplots with consistent styling and a iterator properties.
# We can jump to the next subplot axis using axm.next(), or we can navigate to a specific plot using numpy-like indexing, e.g. axm[0, 1] for the first row and second column.
fig, axm = apt.pl.create_figure(2, 2, figsize=(12, 6), gridspec_kwargs={"width_ratios": [0.5, 3]})
# Define color dictionary for the different treatments
color_dict = {
"CTRL": apt.pl.BaseColors.get("red"),
"Stauro": apt.pl.BaseColors.get("blue"),
}
# Map the sample names to their respective colors based on treatment
sample_color_dict = {
sample: color_dict[treatment]
for sample, treatment in zip(adata_protein.obs_names, adata_protein.obs["treatment"], strict=False)
}
# Iterative plotting over both modalities, convenient to keep them in a dict and iterate over
plot_data = {
"protein": adata_protein,
"precursor": adata_precursor,
}
for readout, adata in plot_data.items():
# Jump to the first subplot for the CV plots
ax = (
axm.next()
) # note that 'ax' is a vanilla matplotlib axis object, so we can call any suitable matplotlib function on it
ax.set_ylim(0, 1)
apt.pl.violinplot(
ax=ax,
data=adata.varm["cv_treatment"],
direct_columns=["CTRL", "Stauro"],
color_dict=color_dict,
)
apt.pl.label_axes(
ax=ax,
xlabel="Treatment",
ylabel=f"{readout.capitalize()} CV",
title=f"{readout.capitalize()} CVs by treatment",
)
# Second subplot for the boxplots of log2 intensities by sample
ax = axm.next()
apt.pl.boxplot(
ax=ax,
data=adata.T,
direct_columns=adata.obs_names.tolist(),
color_dict=sample_color_dict,
)
# Modify ax to process x ticks to fit the plot better
xtick_labels = [item.get_text() for item in ax.get_xticklabels()] # Get xtick labels directly from axes
xtick_labels = [
"_".join(x.split("_")[-2:]) for x in xtick_labels
] # Shorten the xtick labels to make them more readable
_ = ax.set_xticklabels(xtick_labels) # Set the new xtick labels
apt.pl.label_axes(
ax=ax,
xlabel="Sample",
ylabel=f"{readout.capitalize()} log2 intensity",
title=f"{readout.capitalize()} log2 intensities by sample",
)
# save figure
apt.pl.save_figure(
fig=fig, # Note that here we're using the whole figure to whose axes we added our plots
filename="pelsa_quantities_overview.svg",
output_dir="./example_outputs",
)
3. Perform differential expression analysis on precursors#
Peptides from the stablized protein regions are expected to appear downregulated. We perform comparative differential expression analysis:
On the one hand, we evaluate “vanilla” independent two-sample t-test.
On the other, we employ
alphapepttools’ wrapper ofAlphaQuant[2], a differential expression method specially adapted for mass-spectrometry proteomics data.Additionally, we examine a Python-implementation of the popular Limma package’s Empirical Bayes moderated ttest, which is more sophisticated than a simple ttest but considerably faster and more lightweight than alphaquant.
We want to retain the original AnnData object’s feature-level metadata in our differential expression results, which can be accomplished by joining the var object to the results.
3.1 Considerations for missing values#
in practice, differential expression analyses face challenges from missing values: on one hand, losing too many replicates means a feature is dropped and lost to follow up analyses. On the other hand, if imputation is used, features that may be noisy or spurious end up in the final results, which is especially problematic for low input experiments with an enrichment setup: in these experiments, a feature may be expected to drop completely in treatment conditions (e.g. pulldown enrichment or genomic knockout), which would justify imputation of missing values with some kind of baseline. However, if intensities approach the limit of detection (LOD) of an instrument, features may also be missing not at random (MNAR) because of low abundance. Blanket imputation brings back “the good and the bad”: both downregulated and lowly abundant, highly variable features.
in practice, whether or not to impute depends on the experimental setup and the question asked from the data. For PELSA, the standard practice of filtering for 100 % data completeness has emerged as a practical way to reduce false positives, at the cost of some true regulated hits. More sophisticated imputation methods that aim to model missingness[3] may help alleviate this tradeoff in future analyses
comparison = ("Stauro", "CTRL")
3.2 Two-sample independent two-sided t-test#
# Vanilla ttest on peptide data
ttest_peptide_results = apt.tl.diff_exp_ttest(
adata=adata_precursor,
between_column="treatment",
comparison=comparison,
min_valid_values=4, # this implementation of ttest allows for setting a minimum amount of required valid values within each group
equal_var=False,
)
ttest_peptide_results = ttest_peptide_results.join(adata_precursor.var, how="left")
Comparing groups: Stauro vs CTRL
3.3 Two-sample independent two-sided Empirical Bayes Moderated t-test#
# Run Limma Empirical Bayes moderated ttest on peptides: alphapepttools inmoose
# This implementation requires 100 % data completeness, i.e. 4/4 values present per feature per condition
ebayes_peptide_results = apt.tl.diff_exp_ebayes(
adata=adata_precursor,
between_column="treatment",
comparison=comparison,
)[1]
ebayes_peptide_results = ebayes_peptide_results.join(adata_precursor.var, how="left")
3.4 Tree-based differential quantification#
# AlphaQuant ttest on full search engine output (takes approx. 3-4 minutes on a M2 MacBook Pro. Set "REPROCESS" flag to True to re-run, otherwise cached data will be used)
REPROCESS = False
if REPROCESS:
comparison_key, alphaquant_results = apt.tl.diff_exp_alphaquant(
adata=adata_precursor,
report=full_report,
between_column="treatment",
comparison=comparison,
valid_values_filter_mode="either",
min_valid_values=3,
plots="hide",
)
alphaquant_peptide_results = alphaquant_results["peptide"]
alphaquant_peptide_results.to_pickle("./../example_data/notebook_02_alphaquant_peptide_results.pkl")
else:
logger.info(" --> Using cached AlphaQuant results")
alphaquant_peptide_results = pd.read_pickle("./../example_data/notebook_02_alphaquant_peptide_results.pkl")
Alphaquant results are saved into a dictionary based on the quantification level:
results
protein level results
proteoform level results
peptide level results
3.5 Custom precursor aggregation#
In order to go from precursor to protein-level annotation, we perform an aggregation step similar to the original publication: For each protein, the peptide with the highest significance is assigned its gene name. This effectively converts a dataset of precursors into a dataset of unique genes. We perform the same aggregation in our preprocessing here by implementing a custom aggregation function.
def top_peptides_to_proteins(
ttest_df: pd.DataFrame,
y_score_column: str = "-log10(p_value)",
id_column: str = "protein",
) -> pd.DataFrame:
"""Convert peptide level t-test results to protein level by taking the top peptide per protein."""
ttest_df = ttest_df.copy()
return ttest_df.sort_values(y_score_column, ascending=False).groupby(id_column).first().reset_index()
aq_protein_table = top_peptides_to_proteins(alphaquant_peptide_results, id_column="protein")
eb_protein_table = top_peptides_to_proteins(ebayes_peptide_results, id_column="genes")
tt_protein_table = top_peptides_to_proteins(ttest_peptide_results, id_column="genes")
3.6 Kinase annotation#
Since Staurosporine is a pan-kinase inhibitor and we are interested in recovering its targets, we have to annotate all kinases in the data in order to visualize them and perform kinase enrichment scoring. Therefore, we load a table from Gerard et al. [4] (http://www.kinhub.org/kinases.html) and transfer the gene-level annotations to our data.
def annotate_kinases(
ttest_df: pd.DataFrame,
kinase_table_path: str,
gene_id_column: str = "gene",
) -> list:
"""Assign str value for kinase/no kinase based on publication data:
Manning, Gerard, et al. "The protein kinase complement of the human genome." Science 298.5600 (2002): 1912-1934.,
table from: http://www.kinhub.org/kinases.html
"""
ttest_df = ttest_df.copy()
kinase_table = pd.read_excel(kinase_table_path)
kinase_list = kinase_table["HGNC Name"].tolist()
kinase_dict = dict(zip(kinase_list, ["kinase"] * len(kinase_list), strict=False))
ttest_df["kinase_status"] = ttest_df[gene_id_column].map(kinase_dict).fillna("other")
return ttest_df
# Download the kinase table using the alphapepttools data module
kinase_path = apt.data.get_data("kinase_table")
tt_protein_table = annotate_kinases(tt_protein_table, kinase_path, "genes")
eb_protein_table = annotate_kinases(eb_protein_table, kinase_path, "genes")
aq_protein_table = annotate_kinases(aq_protein_table, kinase_path, "protein")
/Users/vincenthbrennsteiner/Documents/mann_labs/_git_repositories/alphapepttools/docs/notebooks/studies/kinome_table.xlsx already exists (0.06418609619140625 MB)
3.7 Custom Kinase Enrichment Score:#
In the main publication [1] and its follow-up [4], a kinase score is implemented to assess the sensitivity and specificity of the assay. This score derives from the fact that, in the ideal case we should only see kinase peptides being regulated by treatment with a kinase-binder. In practice, there are off-targets, i.e. peptides/proteins that appear as downregulated even though they are not kinases. The score is the ratio of downregulated (log2fc < 0) kinases/non-kinases at 80 % specificity when ranking by -log10(p_value).
3.7.1 Kinase Score intuition#
Imagine a boundary moving down from the highest point in the left half of the volcanoplot. Each time the boundary passes a point, we record if the point is a kinase or a non-kinase. When the ratio of passed kinases / passed non-kinases becomes smaller than 0.8 we state that 80 % specificity is reached. However many kinases we have up to this point is our score.
def kinase_score(
ttest_df: pd.DataFrame,
id_col: str,
score_column: str = "-log10(p_value)",
max_candidates: int = 500,
threshold: float = 0.8,
) -> dict | None:
"""Perform kinase scoring analysis per HT-Pelsa.
1. Sort peptides by -log10(significance) (score_column) descending
2. Consider only regulated peptides, i.e. log2fc < 0
3. Iterate through ranks, calculating the cumulative kinase fraction
(number of kinases / total number of peptides)
4. Stop when kinase fraction drops below min_kinase_pct or max_candidates is reached
"""
ttest_df = ttest_df.copy()
print(
f"Total unique kinases in the dataset: {ttest_df.loc[ttest_df['kinase_status'] == 'kinase', id_col].nunique()}"
)
# Restrict to downregulated peptides
ttest_df = ttest_df[ttest_df["log2fc"] < 0]
print(
f"Total unique kinases in the downregulated dataset: {ttest_df.loc[ttest_df['kinase_status'] == 'kinase', id_col].nunique()}"
)
# Rank peptides by score
ttest_df = ttest_df.sort_values(score_column, ascending=False).reset_index(drop=True)
ttest_df["rank"] = ttest_df.index + 1
# Iterate through ranks and record the kinase fraction
def _kinase_fraction(df: pd.DataFrame) -> float:
kinase_count = df[df["kinase_status"] == "kinase"].shape[0]
total_count = df.shape[0]
return kinase_count / total_count if total_count > 0 else np.nan
kinase_fractions = []
for n in range(1, len(ttest_df) + 1):
subset = ttest_df.head(n)
kinase_fraction = _kinase_fraction(subset)
score_threshold = subset[score_column].iloc[-1]
kinase_fractions.append(
{
"rank": n,
"kinase_fraction": kinase_fraction,
"score_threshold": score_threshold,
"n_kinases": subset[subset["kinase_status"] == "kinase"].shape[0],
"n_total": subset.shape[0],
}
)
if n >= max_candidates and kinase_fraction < threshold:
break
kinase_fraction_df = pd.DataFrame(kinase_fractions)
# Determine score based on specificity threshold
above_threshold = kinase_fraction_df[kinase_fraction_df["kinase_fraction"] >= threshold]
optimal_row = above_threshold.iloc[-1] if not above_threshold.empty else None
if optimal_row is None:
print("No suitable threshold found.")
return None
return {
"threshold": optimal_row["score_threshold"],
"n_candidates": optimal_row["rank"],
"n_kinases": optimal_row["n_kinases"],
"n_non_kinases": optimal_row["n_total"] - optimal_row["n_kinases"],
"kinase_percentage": optimal_row["kinase_fraction"],
}
3.8 Volcano plot of differentially regulated genes#
alphapepttools offers a layered, matplotlib-based plotting syntax that allows for quick and flexible generation of volcanoplots, or any kind of layered plot in general (see tutorials/tutorial_02_basic_plotting_workflows.ipynb for reference). The key to this kind of visualization are hierarchical layers that specify which points should be plotted on top, specified by the layers argument of alphapepttools.pl.volcano.
# As before, we establish uniform coloring across all plots
color_dict = {
"kinase": apt.pl.BaseColors.get("red"),
"other": apt.pl.BaseColors.get("grey"),
}
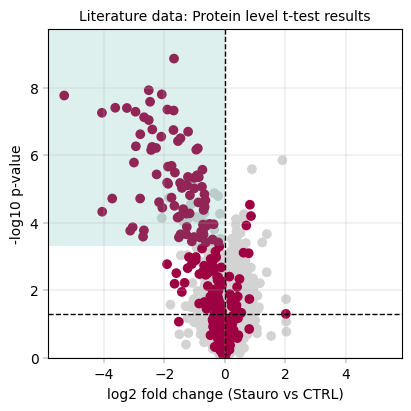
3.8.1 t-test results + kinase scoring#
# Computing the kinase score at a set threshold & small message to summarize the results
ttest_ks_result = kinase_score(tt_protein_table, id_col="genes", threshold=0.8)
print(
f"Kinase scoring threshold at -log10(p_value) = {ttest_ks_result['threshold']:.2f}, covering {ttest_ks_result['n_candidates']} proteins with {ttest_ks_result['n_kinases']} kinases ({ttest_ks_result['kinase_percentage'] * 100:.1f}%)"
)
fig, axm = apt.pl.create_figure(1, 1, figsize=(4, 4))
ax = axm.next()
apt.pl.volcano(
ax=ax,
data=tt_protein_table,
x_column="log2fc",
y_column="-log10(p_value)",
color_dict=color_dict,
layers=[
("kinase_status", "kinase", "kinase"), # column, filter value, color dict key of top layer
("kinase_status", "other", "other"), # column, filter value, color dict key of second layer
],
x_thresholds=(0),
)
apt.pl.label_axes(
ax=ax,
xlabel="log2 fold change (Stauro vs CTRL)",
ylabel="-log10 p-value",
title="Literature data: Protein level t-test results",
)
# For better visualization, we draw a rectangle to indicate the kinase scoring threshold
left_bound = ax.get_xlim()[0]
upper_bound = ax.get_ylim()[1]
rect = Rectangle(
xy=(
left_bound,
ttest_ks_result["threshold"],
), # Bottom-left corner: x value is the left bound of the plot, y value is the kinase scoring threshold
width=np.abs(0 - left_bound), # Width from the left bound to 0, indicating that we focus on downregulated ids
height=upper_bound - ttest_ks_result["threshold"], # Height to figure top
linewidth=2,
facecolor=apt.pl.BaseColors.get("green", alpha=0.2),
)
ax.add_patch(rect)
# save figure
apt.pl.save_figure(
fig=fig, # Note that here we're using the whole figure to whose axes we added our plots
filename="Literature_PELSA_TTEST_AggProt_Stauro.svg",
output_dir="./example_outputs",
)
Total unique kinases in the dataset: 276
Total unique kinases in the downregulated dataset: 212
Kinase scoring threshold at -log10(p_value) = 3.31, covering 105.0 proteins with 84.0 kinases (80.0%)
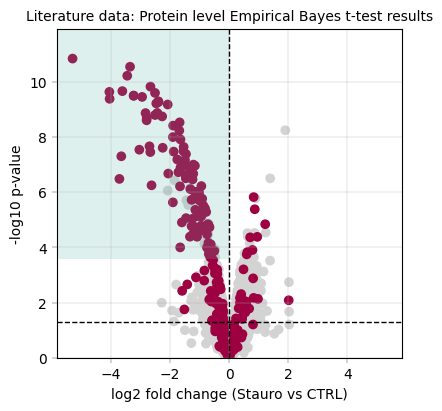
3.8.2 Empirical Bayes moderated ttest (Limma-like) results + kinase scoring#
# Computing the kinase score at a set threshold & small message to summarize the results
ebayes_ttest_ks_results = kinase_score(eb_protein_table, id_col="genes", threshold=0.8)
print(
f"Kinase scoring threshold at -log10(p_value) = {ebayes_ttest_ks_results['threshold']:.2f}, covering {ebayes_ttest_ks_results['n_candidates']} proteins with {ebayes_ttest_ks_results['n_kinases']} kinases ({ebayes_ttest_ks_results['kinase_percentage'] * 100:.1f}%)"
)
fig, axm = apt.pl.create_figure(1, 1, figsize=(4, 4))
ax = axm.next()
apt.pl.volcano(
ax=ax,
data=eb_protein_table,
x_column="log2fc",
y_column="-log10(p_value)",
color_dict=color_dict,
layers=[
("kinase_status", "kinase", "kinase"), # column, filter value, color name
("kinase_status", "other", "other"),
],
x_thresholds=(0),
)
apt.pl.label_axes(
ax=ax,
xlabel="log2 fold change (Stauro vs CTRL)",
ylabel="-log10 p-value",
title="Literature data: Protein level Empirical Bayes t-test results",
)
# For better visualization, we draw a rectangle to indicate the kinase scoring threshold
left_bound = ax.get_xlim()[0]
upper_bound = ax.get_ylim()[1]
rect = Rectangle(
xy=(
left_bound,
ebayes_ttest_ks_results["threshold"],
), # Bottom-left corner: x value is the left bound of the plot, y value is the kinase scoring threshold
width=np.abs(0 - left_bound), # Width from the left bound to 0, indicating that we focus on downregulated ids
height=upper_bound - ebayes_ttest_ks_results["threshold"], # Height to figure top
linewidth=2,
facecolor=apt.pl.BaseColors.get("green", alpha=0.2),
)
ax.add_patch(rect)
# save figure
apt.pl.save_figure(
fig=fig, # Note that here we're using the whole figure to whose axes we added our plots
filename="Literature_PELSA_EBAYES_TTEST_AggProt_Stauro.svg",
output_dir="./example_outputs",
)
Total unique kinases in the dataset: 253
Total unique kinases in the downregulated dataset: 197
Kinase scoring threshold at -log10(p_value) = 3.57, covering 117.0 proteins with 94.0 kinases (80.3%)
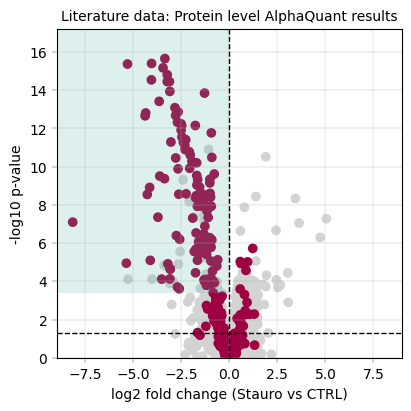
3.8.3 Alphaquant results + kinase scoring#
# Computing the kinase score at a set threshold & small message to summarize the results
aq_ks_result = kinase_score(aq_protein_table, id_col="protein", threshold=0.8)
print(
f"Kinase scoring threshold at -log10(p_value) = {aq_ks_result['threshold']:.2f}, covering {aq_ks_result['n_candidates']} proteins with {aq_ks_result['n_kinases']} kinases ({aq_ks_result['kinase_percentage'] * 100:.1f}%)"
)
fig, axm = apt.pl.create_figure(1, 1, figsize=(4, 4))
ax = axm.next()
apt.pl.volcano(
ax=ax,
data=aq_protein_table,
x_column="log2fc",
y_column="-log10(p_value)",
color_dict=color_dict,
layers=[
("kinase_status", "kinase", "kinase"), # column, filter value, color name
("kinase_status", "other", "other"),
],
x_thresholds=(0),
)
apt.pl.label_axes(
ax=ax,
xlabel="log2 fold change (Stauro vs CTRL)",
ylabel="-log10 p-value",
title="Literature data: Protein level AlphaQuant results",
)
# For better visualization, we draw a rectangle to indicate the kinase scoring threshold
left_bound = ax.get_xlim()[0]
upper_bound = ax.get_ylim()[1]
rect = Rectangle(
xy=(
left_bound,
aq_ks_result["threshold"],
), # Bottom-left corner: x value is the left bound of the plot, y value is the kinase scoring threshold
width=np.abs(0 - left_bound), # Width from the left bound to 0, indicating that we focus on downregulated ids
height=upper_bound - aq_ks_result["threshold"], # Height to figure top
linewidth=2,
facecolor=apt.pl.BaseColors.get("green", alpha=0.2),
)
ax.add_patch(rect)
# save figure
apt.pl.save_figure(
fig=fig, # Note that here we're using the whole figure to whose axes we added our plots
filename="Literature_PELSA_AlphaQuant_AggProt_Stauro.svg",
output_dir="./example_outputs",
)
Total unique kinases in the dataset: 276
Total unique kinases in the downregulated dataset: 215
Kinase scoring threshold at -log10(p_value) = 3.40, covering 137.0 proteins with 110.0 kinases (80.3%)
3.9 Visualizing the kinase enrichment results compared to literature data#
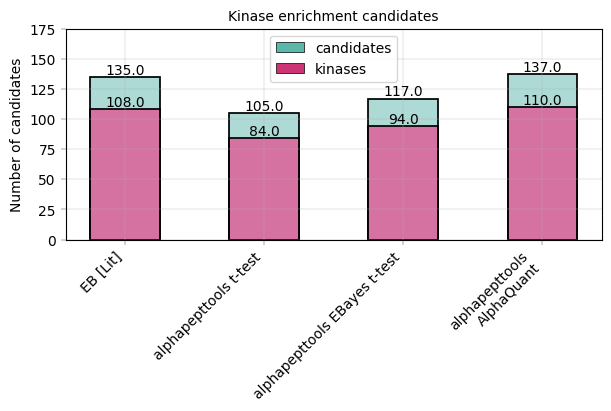
In Figure 2 b of Kejia, Li et al., the number of downregulated vs. kinase proteins at 80 % specificity (as computed above) is given at 135 downregulated candidates and 108 kinase targets. Note that their result were computed using an Empirical Bayes test. We can visualize this data and compare to our alphapepttools differential expression data:
total_candidates = pd.DataFrame(
{
"group": ["EB [Lit]", "alphapepttools t-test", "alphapepttools EBayes t-test", "alphapepttools\nAlphaQuant"],
"value": [
135,
ttest_ks_result["n_candidates"],
ebayes_ttest_ks_results["n_candidates"],
aq_ks_result["n_candidates"],
],
}
)
total_kinases = pd.DataFrame(
{
"group": ["EB [Lit]", "alphapepttools t-test", "alphapepttools EBayes t-test", "alphapepttools\nAlphaQuant"],
"value": [108, ttest_ks_result["n_kinases"], ebayes_ttest_ks_results["n_kinases"], aq_ks_result["n_kinases"]],
}
)
color_dict = {
"candidates": apt.pl.BaseColors.get("green"),
"kinases": apt.pl.BaseColors.get("red", lighten=0.1, alpha=0.8),
}
fig, axm = apt.pl.create_figure(1, 1, figsize=(6, 4))
# First, plot the total candidates
ax = axm.next()
ax.set_ylim(0, 175)
apt.pl.barplot(
ax=ax,
data=total_candidates,
grouping_column="group",
value_column="value",
color=apt.pl.BaseColors.get("green"),
)
total_candidates["position"] = range(len(total_candidates))
apt.pl.label_plot(
ax=ax,
data=total_candidates,
x_column="position",
y_column="value",
label_column="value",
label_kwargs={"va": "bottom", "ha": "center"},
)
# Then, add the kinases
apt.pl.barplot(
ax=ax,
data=total_kinases,
grouping_column="group",
value_column="value",
color=apt.pl.BaseColors.get("red", lighten=0.3),
)
ax.set_xticklabels(ax.get_xticklabels(), rotation=45, ha="right")
total_kinases["position"] = range(len(total_kinases))
apt.pl.label_plot(
ax=ax,
data=total_kinases,
x_column="position",
y_column="value",
label_column="value",
label_kwargs={"va": "bottom", "ha": "center"},
)
apt.pl.label_axes(
ax=ax,
xlabel="",
ylabel="Number of candidates",
title="Kinase enrichment candidates",
)
apt.pl.add_legend_to_axes(
ax=ax,
levels=color_dict,
)
# save figure
apt.pl.save_figure(
fig=fig, # Note that here we're using the whole figure to whose axes we added our plots
filename="Literature_PELSA_Kinase_Enrichment_Candidates_Comparison.svg",
output_dir="./example_outputs",
)
Summary#
In summary, we saw that alphapepttools…
can swiftly load precursor and protein data into
AnnDatainstancescontains standardized, ‘plug and play’ modules for proteomics differential expression
offers strong basic functions but remains easily extendable to custom functions and use-cases, chiefly by
Avoiding end-to-end data processing pipelines, instead keeping Python data structures accessible at all times
Avoiding end-to-end plotting pipelines, instead keeping Matplotlib axes accessible at all times for customization
Note:#
Access to data structures and plot axes is especially useful in the light of LLM-assisted data analysis, where exact syntax knowledge of e.g. Matplotlib is no longer required. Under these aspects, any abstraction (e.g. via a GUI) is liable to be more limiting than standardized Python objects, the manipulation of which is amply covered by current LLMs. We believe that this mix of strong ‘atomic’ functions and custom extensions for a given analysis is ideal to handle modern proteomics data analysis in a reproducible & shareable way.