Basic alphapepttools workflow#
load data and meta data
basic minimal preprocessing
perform PCA
plot PCA embeddings, variance and loadings
import logging
import numpy as np
from pathlib import PureWindowsPath
import pandas as pd
import tempfile
import alphapepttools as at
from alphapepttools.pl.figure import create_figure, label_axes
from alphapepttools.pl.plots import Plots
logging.basicConfig(level=logging.INFO)
Load the data#
We download the data from this folder (ca. 2 megabytes)
output_dir = "./datasets/data_for_04_basic_PCA_workflow"
pg_table_path = at.data.get_data("hela_pg_diann", output_dir=output_dir if output_dir else tempfile.mkdtemp())
metadata_path = at.data.get_data("hela_metadata", output_dir=output_dir if output_dir else tempfile.mkdtemp())
./datasets/data_for_04_basic_PCA_workflow/report.pg_matrix.tsv already exists (1.0532913208007812 MB)
./datasets/data_for_04_basic_PCA_workflow/simple_metadata.csv already exists (0.0013446807861328125 MB)
We can read the data easily with the alphapepttools.io functionalities and match the metadata to the observations
# Read PG table and
# Parse full filepaths to file names (as in metadata file)
# Set uniprot ids as index
adata = at.io.read_pg_table(pg_table_path, search_engine="diann")
adata.obs_names = adata.obs_names.to_series().apply(lambda x: PureWindowsPath(str(x)).stem)
adata.var = adata.var.reset_index(names="protein_names").set_index("uniprot_ids")
# Read metadata
metadata = pd.read_csv(metadata_path, sep=",", index_col="filename")
adata = at.pp.add_metadata(adata=adata, incoming_metadata=metadata, axis=0)
adata
AnnData object with n_obs × n_vars = 18 × 4954
obs: 'replicate', 'fraction'
var: 'protein_names', 'genes', 'description'
Basic EDA on a synthetic example dataset:#
Generate example data
Filter for data completeness on sample level
Visualize samples as histograms
Save data
Filter by data completeness:#
Remove features which have more than the allowed fraction of missing values
print("The numeric data in the anndata object:")
display(adata.to_df().head())
print("The sample-level metadata in the anndata object:")
display(adata.obs.head())
print("The feature-level metadata in the anndata object:")
display(adata.var.head())
# filter out features with more than 25 % missing values
print("Before filtering, the shape of the anndata object: ", adata.shape)
adata = at.pp.filter_data_completeness(adata=adata, max_missing=0.25, action="drop")
print("After filtering, the shape of the anndata object: ", adata.shape)
print("The numeric data in the anndata object:")
display(adata.to_df().head())
print("The sample-level metadata in the anndata object:")
display(adata.obs.head())
print("The feature-level metadata in the anndata object:")
display(adata.var.head())
The numeric data in the anndata object:
The sample-level metadata in the anndata object:
The feature-level metadata in the anndata object:
Before filtering, the shape of the anndata object: (18, 4954)
After filtering, the shape of the anndata object: (18, 2831)
The numeric data in the anndata object:
The sample-level metadata in the anndata object:
The feature-level metadata in the anndata object:
| uniprot_ids | A0A024R1R8;Q9Y2S6 | A0A0B4J2F0 | A0A0J9YX75;A0A0J9YXY3;P0DPF7 | A0AV96 | A0FGR8 | A0JLT2 | A0PJW6 | A1L0T0 | A1L390 | A1X283 | ... | Q9Y6N5 | Q9Y6N6 | Q9Y6N7 | Q9Y6N8 | Q9Y6R0 | Q9Y6V7 | Q9Y6W5 | Q9Y6X3 | Q9Y6X9 | Q9Y6Y8 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5_Map2_3K | 9506220.0 | 9930580.0 | NaN | 6414570.0 | 29546900.0 | NaN | 19563300.0 | 23059400.0 | 1346120.0 | 11691800.0 | ... | 39051400.0 | 172419.0 | 6796840.0 | 8998980.0 | NaN | NaN | 5111880.0 | NaN | NaN | 7060290.0 |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_1K_20210131142601 | 25866000.0 | NaN | NaN | 18604800.0 | 18958500.0 | 3202770.0 | NaN | 14090600.0 | 790375.0 | 23128600.0 | ... | 10291200.0 | NaN | 3829290.0 | NaN | 574729.0 | 19405500.0 | 6310680.0 | 10867000.0 | 11684000.0 | 13398300.0 |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_3K | 10150200.0 | 11627800.0 | NaN | 6550420.0 | 28679800.0 | NaN | 19387300.0 | 24248100.0 | 963907.0 | 9263040.0 | ... | 38928800.0 | NaN | 5441930.0 | 9034600.0 | 1026870.0 | NaN | 3963610.0 | NaN | NaN | 6189950.0 |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_6K | 14660600.0 | 8741560.0 | NaN | 9638120.0 | 35471700.0 | NaN | 15716300.0 | 36269500.0 | NaN | 11779400.0 | ... | 33905400.0 | 237138.0 | 5068170.0 | 7413300.0 | 965536.0 | NaN | 4858220.0 | NaN | NaN | 6046510.0 |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_12K | 23477900.0 | 1497570.0 | NaN | 16496400.0 | 27413400.0 | NaN | 2334540.0 | 24950500.0 | NaN | 21687600.0 | ... | 8834620.0 | 187094.0 | 3736040.0 | 4581280.0 | 1516620.0 | NaN | 7919380.0 | NaN | NaN | 9935580.0 |
5 rows × 4954 columns
| replicate | fraction | |
|---|---|---|
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5_Map2_3K | Map2 | 3K |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_1K_20210131142601 | Map1 | 1K |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_3K | Map1 | 3K |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_6K | Map1 | 6K |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_12K | Map1 | 12K |
| protein_names | genes | description | |
|---|---|---|---|
| uniprot_ids | |||
| A0A024R1R8;Q9Y2S6 | TMA7B_HUMAN;TMA7_HUMAN | TMA7;TMA7B | Translation machinery-associated protein 7B |
| A0A0B4J2F0 | PIOS1_HUMAN | PIGBOS1 | Protein PIGBOS1 |
| A0A0J9YX75;A0A0J9YXY3;P0DPF7 | TVB62_HUMAN;TVB63_HUMAN;TVB69_HUMAN | TRBV6-2;TRBV6-3;TRBV6-9 | T cell receptor beta variable 6-9 |
| A0AV96 | RBM47_HUMAN | RBM47 | RNA-binding protein 47 |
| A0FGR8 | ESYT2_HUMAN | ESYT2 | Extended synaptotagmin-2 |
| uniprot_ids | A0A024R1R8;Q9Y2S6 | A0AV96 | A0FGR8 | A1L0T0 | A1X283 | A5PLL7 | A5YKK6 | A6NCE7;Q9GZQ8 | A6NDG6 | A6NHR9 | ... | Q9Y6K5 | Q9Y6M0 | Q9Y6M1 | Q9Y6M4 | Q9Y6M5 | Q9Y6N7 | Q9Y6N8 | Q9Y6R0 | Q9Y6W5 | Q9Y6Y8 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5_Map2_3K | 9506220.0 | 6414570.0 | 29546900.0 | 23059400.0 | 11691800.0 | 2741150.0 | 10835800.0 | 5629360.0 | 561137.0 | 3789200.0 | ... | 8955980.0 | 27799800.0 | 3070990.0 | 3151630.0 | 64329500.0 | 6796840.0 | 8998980.0 | NaN | 5111880.0 | 7060290.0 |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_1K_20210131142601 | 25866000.0 | 18604800.0 | 18958500.0 | 14090600.0 | 23128600.0 | 4624200.0 | 11243600.0 | 7728880.0 | 462468.0 | 47274600.0 | ... | 9021290.0 | 18298000.0 | NaN | 1579620.0 | 38458300.0 | 3829290.0 | NaN | 574729.0 | 6310680.0 | 13398300.0 |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_3K | 10150200.0 | 6550420.0 | 28679800.0 | 24248100.0 | 9263040.0 | 3809220.0 | 10480100.0 | 7059070.0 | 700098.0 | NaN | ... | 7307580.0 | 23668700.0 | 3084110.0 | 3130200.0 | 60378200.0 | 5441930.0 | 9034600.0 | 1026870.0 | 3963610.0 | 6189950.0 |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_6K | 14660600.0 | 9638120.0 | 35471700.0 | 36269500.0 | 11779400.0 | 5127720.0 | 19063600.0 | 8905940.0 | 582714.0 | NaN | ... | 12709900.0 | 39207800.0 | 6057120.0 | 3672700.0 | 56567800.0 | 5068170.0 | 7413300.0 | 965536.0 | 4858220.0 | 6046510.0 |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_12K | 23477900.0 | 16496400.0 | 27413400.0 | 24950500.0 | 21687600.0 | 5740560.0 | 39849800.0 | 8597510.0 | 595144.0 | 3586450.0 | ... | 18124000.0 | 30144100.0 | 10509100.0 | 3353660.0 | 42608500.0 | 3736040.0 | 4581280.0 | 1516620.0 | 7919380.0 | 9935580.0 |
5 rows × 2831 columns
| replicate | fraction | |
|---|---|---|
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5_Map2_3K | Map2 | 3K |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_1K_20210131142601 | Map1 | 1K |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_3K | Map1 | 3K |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_6K | Map1 | 6K |
| 20210131_EXPL4_ViAl_SA_HeLa_Evosep_21min_DIA_120k_15k_1-5s_Map1_12K | Map1 | 12K |
| protein_names | genes | description | |
|---|---|---|---|
| uniprot_ids | |||
| A0A024R1R8;Q9Y2S6 | TMA7B_HUMAN;TMA7_HUMAN | TMA7;TMA7B | Translation machinery-associated protein 7B |
| A0AV96 | RBM47_HUMAN | RBM47 | RNA-binding protein 47 |
| A0FGR8 | ESYT2_HUMAN | ESYT2 | Extended synaptotagmin-2 |
| A1L0T0 | HACL2_HUMAN | ILVBL | 2-hydroxyacyl-CoA lyase 2 |
| A1X283 | SPD2B_HUMAN | SH3PXD2B | SH3 and PX domain-containing protein 2B |
Creating new layers prior to preprocessing#
This way, we can save the raw data and try different pp steps on the raw data.
# save the raw data before log transformation
adata.layers["raw"] = adata.X.copy()
# log2 transform the data
adata.X = np.log2(adata.X + 1)
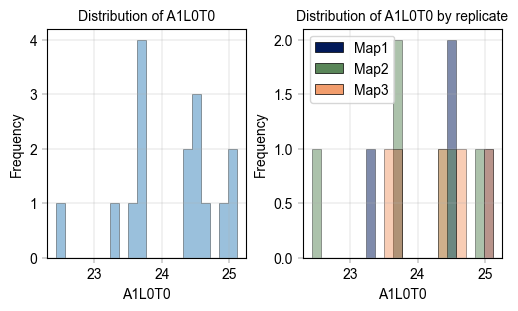
Visualize the distribution of values in different levels of an observational metadata variable#
In this example, check the distribution of “gene_1” expression values per cell type.
# Apply the AxisManager to make axes iterable and apply consistent alphapepttools styling.
# Axes can also be accessed directly by indexing the axm object.
fig, axm = create_figure(nrows=1, ncols=2, figsize=(5, 3))
# Plot.histogram handles adata natively. Columns from the data and metadata are accessible
# Focus on the distribution of protein A1L0T0
ax = axm.next()
Plots.histogram(
data=adata,
value_column="A1L0T0",
bins=20,
legend="auto",
ax=ax,
hist_kwargs={"alpha": 0.5, "histtype": "stepfilled", "linewidth": 0.5, "edgecolor": "black"},
)
label_axes(ax, "A1L0T0", "Frequency", "Distribution of A1L0T0")
# Focus on the distribution of protein A1L0T0 in the different replicates
ax = axm.next()
Plots.histogram(
data=adata,
value_column="A1L0T0",
color_map_column="replicate",
bins=20,
legend="auto",
ax=ax,
hist_kwargs={"alpha": 0.5, "histtype": "stepfilled", "linewidth": 0.5, "edgecolor": "black"},
)
label_axes(ax, "A1L0T0", "Frequency", "Distribution of A1L0T0 by replicate")
# # save figure
# save_figure(
# fig=fig,
# filename="sample_histogram.png",
# output_dir=output_directory,
# dpi=300,
# transparent=False,
# )
Running PCA#
Before running PCA, we need to filter out NaN values. PCA can not be computed on matrices with missing values. Therefore, prior to PCA, we will create a list of ‘core proteins’ of proteins detected in all observations, save it in the feature meta data frame (adata.var)
# add a new column to the adata.var object with the name "is_core" to indicate whether the feature is part of the core proteome
adata = at.pp.filter_data_completeness(adata, max_missing=0, action="flag", var_colname="is_core")
# view hoe many features are part of the core proteome
print("The number of features in the core proteome:")
print(adata.var["is_core"].value_counts())
The number of features in the core proteome:
is_core
True 1806
False 1025
Name: count, dtype: int64
Now we can run PCA, specifying the adata.var column that filters the proteins by 100% completeness:
# this function is now implemented on sample level (PCA of the observations).
at.tl.pca(adata, meta_data_mask_column_name="is_core", n_comps=10)
# view the PCA results
print("The dimensions of PC coordinates in the adata.obsm are (n_obs x n_comp):")
print(adata.obsm["X_pca_obs"].shape)
print("The PCA loadings in the adata.varm are (n_var x n_comp):")
print(adata.varm["PCs_obs"].shape)
print("Ratio of explained variance (n_comp):")
print(adata.uns["variance_pca_obs"]["variance_ratio"])
print("The explained variance (n_comp):")
print(adata.uns["variance_pca_obs"]["variance"])
The dimensions of PC coordinates in the adata.obsm are (n_obs x n_comp):
(18, 10)
The PCA loadings in the adata.varm are (n_var x n_comp):
(2831, 10)
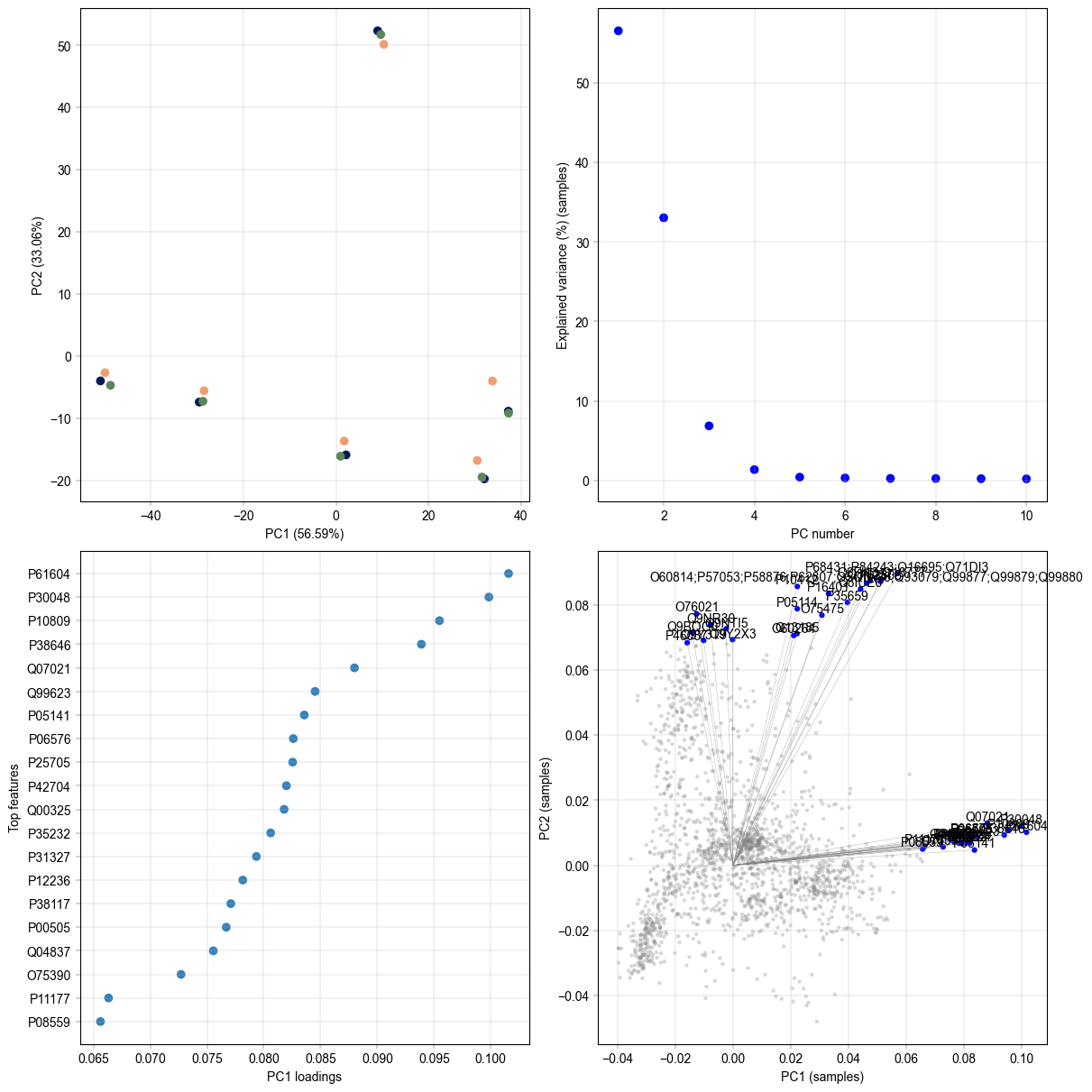
Ratio of explained variance (n_comp):
[0.56593504 0.33056594 0.06844902 0.01339105 0.00388991 0.00288049
0.0023081 0.00220095 0.00194543 0.0017315 ]
The explained variance (n_comp):
[1009.50632405 589.65850994 122.09831939 23.88675892 6.93875425
5.13817412 4.11715411 3.92601867 3.47022027 3.08861607]
In addition to running PCA to get a dimentional reduction of the observations (samples), we can also perform PCA on the features (proteins).
# Now run PCA on the protein space to get their projection in the PCA space.
at.tl.pca(adata, meta_data_mask_column_name="is_core", n_comps=10, dim_space="var")
# view the PCA results for features
print("----- PCA ON FEATURES -----")
print("The dimensions of PC coordinates in the adata.varm are (n_obs x n_comp):")
print(adata.varm["X_pca_var"].shape)
print("The PCA loadings of the samples in the adata.obsm are (n_var x n_comp):")
print(adata.obsm["PCs_var"].shape)
print("Ratio of explained variance (n_comp):")
print(adata.uns["variance_pca_var"]["variance_ratio"])
print("The explained variance (n_comp):")
print(adata.uns["variance_pca_var"]["variance"])
----- PCA ON FEATURES -----
The dimensions of PC coordinates in the adata.varm are (n_obs x n_comp):
(2831, 10)
The PCA loadings of the samples in the adata.obsm are (n_var x n_comp):
(18, 10)
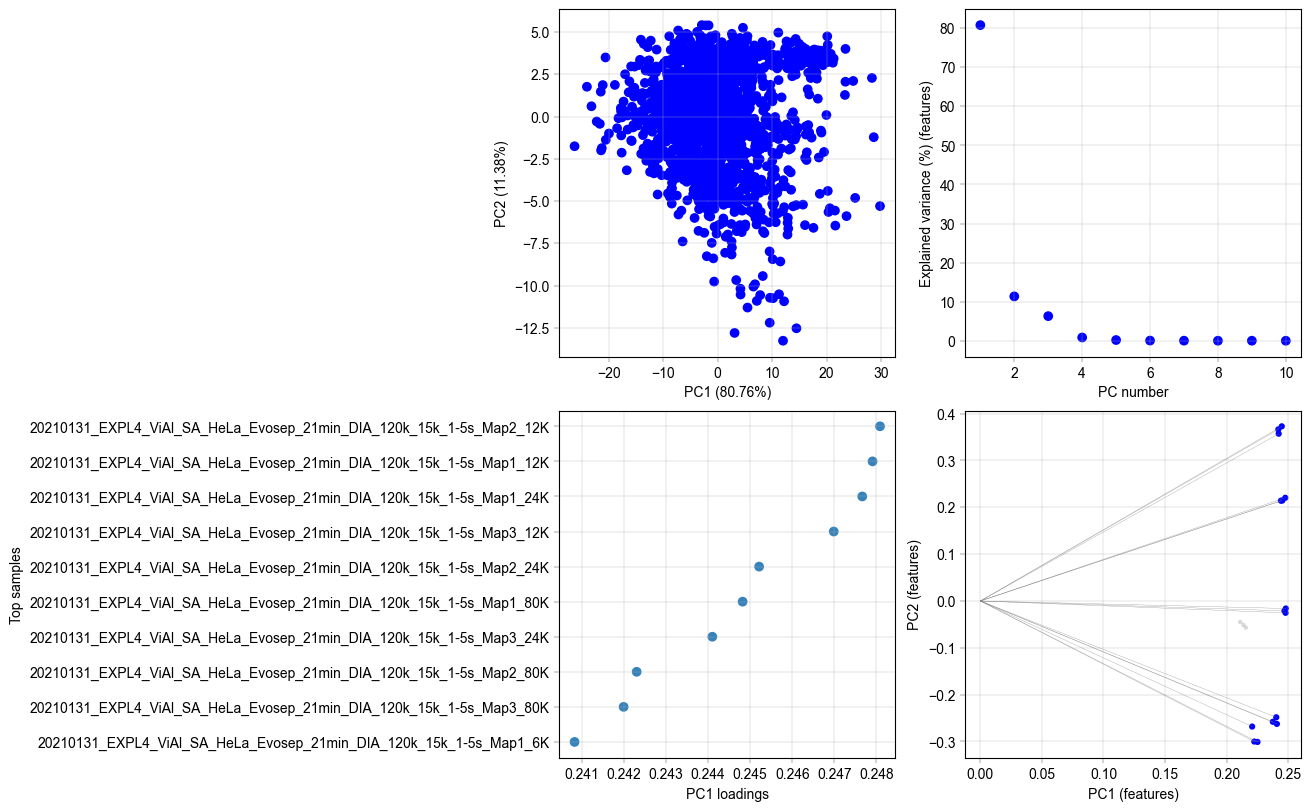
Ratio of explained variance (n_comp):
[8.07567317e-01 1.13818512e-01 6.33157115e-02 8.74927065e-03
2.21018860e-03 7.89962453e-04 5.76627224e-04 4.64255895e-04
4.48561680e-04 3.63473757e-04]
The explained variance (n_comp):
[6.64865141e+01 9.37060716e+00 5.21274308e+00 7.20322002e-01
1.81963451e-01 6.50371166e-02 4.74733601e-02 3.82218986e-02
3.69298036e-02 2.99245680e-02]
Plot PCA results#
We can plot the PCA results on a 2D projection, look at the explained var in each PC using the scree plot, and plot the loadings od the PCs, either per PC or a scatter of 2 PCs, to understand their ‘drivers’.
fig, axm = create_figure(2, 2, figsize=(12, 12))
ax = axm.next()
# PCA plot colored by replicate
Plots.plot_pca(
data=adata,
ax=ax,
x_column=1,
y_column=2,
label=False,
label_column=None,
embeddings_name=None,
color_map_column="replicate",
)
# scree plot to show the explained variance by each PC
ax = axm.next()
Plots.scree_plot(adata=adata, ax=ax, n_pcs=50)
# top loadings of the first PC
ax = axm.next()
Plots.plot_pca_loadings(
data=adata,
ax=ax,
dim=1,
nfeatures=20,
)
# 2d loading plot with highlighted top 20 loadings
ax = axm.next()
Plots.plot_pca_loadings_2d(
data=adata,
ax=ax,
pc_x=1,
pc_y=2,
nfeatures=20,
add_labels=True,
add_lines=True,
scatter_kwargs=None,
)
Plot PCA results for feature PCA#
Just like the PCA on the samples, we can plot the same plots for the results of PCA calculated on the features.
# now produce the PCAs plot for the features
fig, axm = create_figure(2, 2, figsize=(13, 8))
ax = axm.next()
Plots.plot_pca(
data=adata,
ax=ax,
x_column=1,
y_column=2,
dim_space="var",
label=False,
label_column=None,
embeddings_name=None,
)
ax = axm.next()
Plots.scree_plot(adata=adata, ax=ax, n_pcs=50, dim_space="var")
ax = axm.next()
Plots.plot_pca_loadings(data=adata, ax=ax, dim=1, nfeatures=10, dim_space="var")
ax = axm.next()
Plots.plot_pca_loadings_2d(
data=adata,
ax=ax,
pc_x=1,
pc_y=2,
nfeatures=10,
add_labels=False,
add_lines=True,
scatter_kwargs=None,
dim_space="var",
)
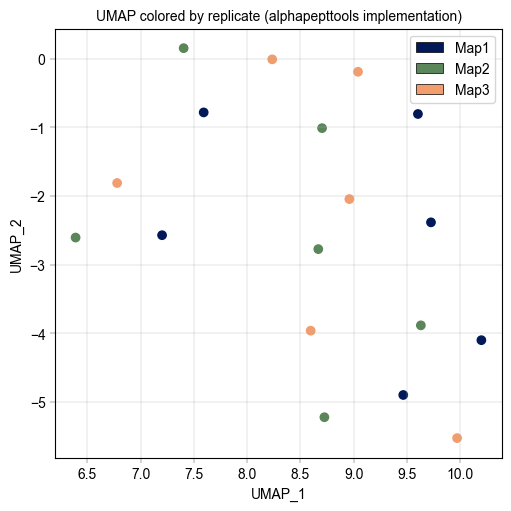
UMAP Visualization with Scanpy#
To explore and visualize the high-dimensional proteomics data, we use UMAP (Uniform Manifold Approximation and Projection) as implemented in Scanpy. UMAP projects complex, high-dimensional feature spaces into a lower-dimensional space (typically 2D) while preserving the local and global structure of the data. This allows us to identify clusters, relationships, and potential outliers in the proteomic profiles at a glance.
In this notebook, Scanpy’s sc.pp.neighbors() and sc.tl.umap() functions are applied to the processed data matrix to compute a nearest-neighbor graph and then generate UMAP coordinates. We will use the sample PCA matrix for neighbor calculations. The resulting UMAP embedding provides an intuitive visualization of sample similarity and grouping based on proteomic features, complementing downstream analyses such as clustering or differential expression.
import scanpy as sc
sc.pp.neighbors(adata, n_neighbors=10, use_rep="X_pca_obs") # use the PCA results on samples
sc.tl.umap(adata)
# location of the umap coordinates in the adata.obsm
print("The UMAP coordinates in the adata.obsm are in adata.obsm['X_umap'] with shape: ", adata.obsm["X_umap"].shape)
print(adata.obsm["X_umap"])
The UMAP coordinates in the adata.obsm are in adata.obsm['X_umap'] with shape: (18, 2)
[[ 7.4034281e+00 1.5592499e-01]
[ 7.2009835e+00 -2.5695477e+00]
[ 7.5915337e+00 -7.8027385e-01]
[ 9.6020727e+00 -8.0324888e-01]
[ 9.7245064e+00 -2.3821766e+00]
[ 1.0197392e+01 -4.0992904e+00]
[ 9.4643431e+00 -4.8978992e+00]
[ 6.3894439e+00 -2.6039217e+00]
[ 8.7024069e+00 -1.0093675e+00]
[ 8.6669664e+00 -2.7722642e+00]
[ 9.6294870e+00 -3.8831391e+00]
[ 8.7243156e+00 -5.2219939e+00]
[ 6.7794323e+00 -1.8091360e+00]
[ 8.2347479e+00 -7.1188253e-03]
[ 9.0395899e+00 -1.8811136e-01]
[ 8.9587097e+00 -2.0429587e+00]
[ 8.5955973e+00 -3.9610267e+00]
[ 9.9694805e+00 -5.5260568e+00]]
Plot UMAP#
We can either plot the UMAP results using scanpy’s plotting function, or we can use alphapepttools plotting function, with adding the umap coordinates directly to the obs df.
# scanpy's plotting function
sc.pl.umap(adata, color="replicate", size=100) # the size is usually much smaller
Another option is to copy the coordinates into the adata.obs data frame, to plot in using scatter function in alphapepttools package
adata.obs["UMAP1"] = adata.obsm["X_umap"][:, 0]
adata.obs["UMAP2"] = adata.obsm["X_umap"][:, 1]
fig, axm = create_figure(1, 1, figsize=(5, 5))
ax = axm.next()
Plots.scatter(adata, x_column="UMAP1", y_column="UMAP2", color_map_column="replicate", ax=ax, legend="auto")
label_axes(
ax=ax,
xlabel="UMAP_1",
ylabel="UMAP_2",
title="UMAP colored by replicate (alphapepttools implementation)",
)